
Software
eSBMTools
eSBMTools is a Python 2.7 package that assists in the setup and evaluation of structure-based simulations of proteins and nucleic acids, both at Cα and all-atom level. The tools interface with GROMACS and support its standard output formats. Standard structure-based models can easily be complemented by information from other sources, like bioinformatics or experimental data.
eSBMTools is a powerful package that makes the job of running simulations much easier. It can be used to generate the necessary input simulation files for GROMACS, compile data from several simulations, produce free energy graphs, make histograms, and a lot more.
The tools are well documented so that you can write your own Python scripts for more complex problems and an overall better control over the simulation. To help you get a feel of what eSBMTools can do, a few examples have been compiled and are available in the package itself.
See also Lutz, B., Sinner, C., Heuermann, G., Verma, A., & Schug, A. (2013). eSBMTools 1.0: enhanced native structure-based modeling tools. Bioinformatics, 29(21), 2795-2796.
diSTruct
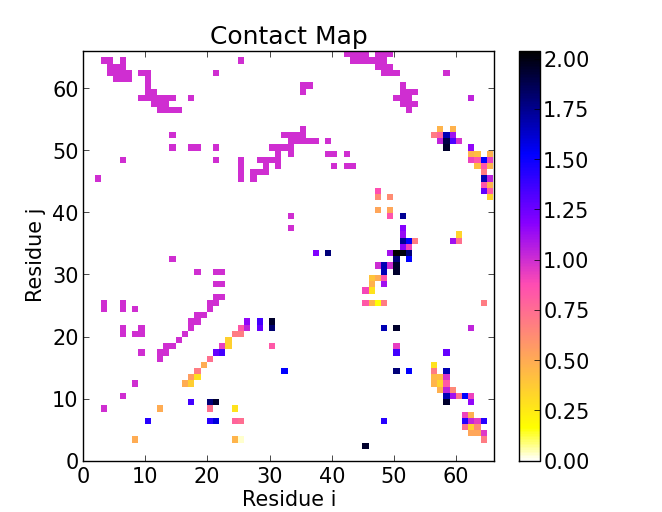
diSTruct is a python package to generate 3D molecular structures from distance constraints. The distance geometry problem is often encountered in molecular biology and the life sciences at large as a host of experimental methods produce ambiguous and noisy distance data. We present diSTruct, an adaptation of the generic MaxEnt-Stress graph drawing algorithm to the domain of biological macromolecules. diSTruct is fast, provides reliable structural models even from incomplete or noisy distance data, and integrates access to graph analysis tools. diSTruct is written in C++, Cython, and Python 3. It is available from GitHub or in the Python package index under the MIT license.
We directly solve the distance geometry problem for arbitrary distance data from experimental or other sources. The key achievements are speed of interpretation, minimizing the error with respect to constraints, and the ability to deal with sparse and noisy data. The rapid construction of 3D models even for a large protein of 700 amino acids takes only on the order of 10 s on common CPUs. This allows to interactively interpret possible sources of ambiguity or errors in the input distance data and helps to improve, e.g., NMR shift assignment.
The algorithm is implemented in the python package diSTruct. diSTruct builds on Biopython (Cock et al., 2009; Hamelryck and Manderick, 2003), a toolkit for computational biology and the NetworKit (Staudt et al., 2016) package for graph analysis.
Our design focus is to provide an interactive Python-based toolkit that
- provides structural models from distance data with minimal constraint errors,
- is able to handle noisy and incomplete data,
- maintains familiarity for users that know Biopython, and
- provides an interface from biological context to a rich graph analysis suite.
See also Taubert, O., Reinartz, I., Meyerhenke, H., & Schug, A. (2019). diSTruct v1. 0: generating biomolecular structures from distance constraints. Bioinformatics, 35(24), 5337-5338.
FLAPS
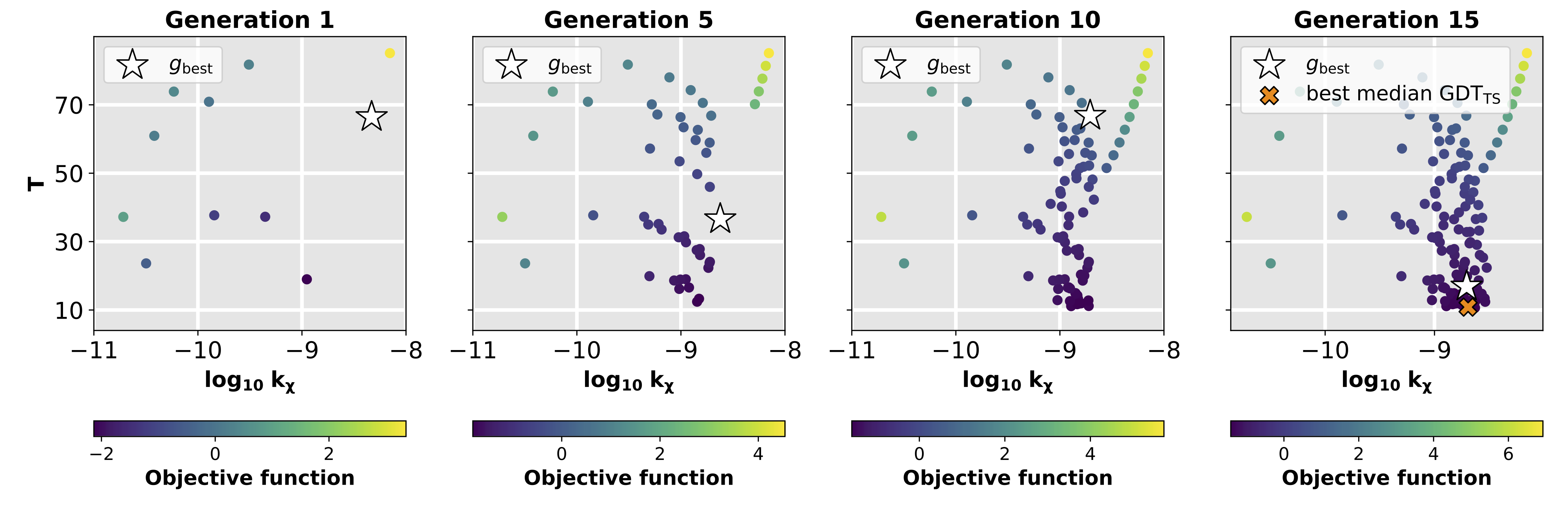
FLAPS (Flexible Self-Adapting Particle Swarm Optimization) is a self-adapting, swarm-intelligence based optimizer that solves the problem of weighting various quality features in multi-response optimization problems. The algorithm is suited for optimizing composite objective functions that depend on both the optimization parameters of interest and additional, a priori unknown weighting paramters which significantly influence the search-space topology. These weighting parameters are learned at runtime, yielding a dynamically evolving and iteratively refined search-space topology. FLAPS is implemented in python and publicly available on GitHub.
We applied FLAPS to the metaheuristic optimization of MD parameters for data-assisted protein simulations. Such simulations are a powerful tool to interpret ambiguous experimental data for structural models. Their performance crucially depends on the non-trivial choice of MD parameters, where the key challenge is balancing experimental information and the physical model. Using a specifically designed objective function, we showed FLAPS to be capable of finding functional MD parameters for SAXS-guided structure-based simulations of two proteins.
pydca
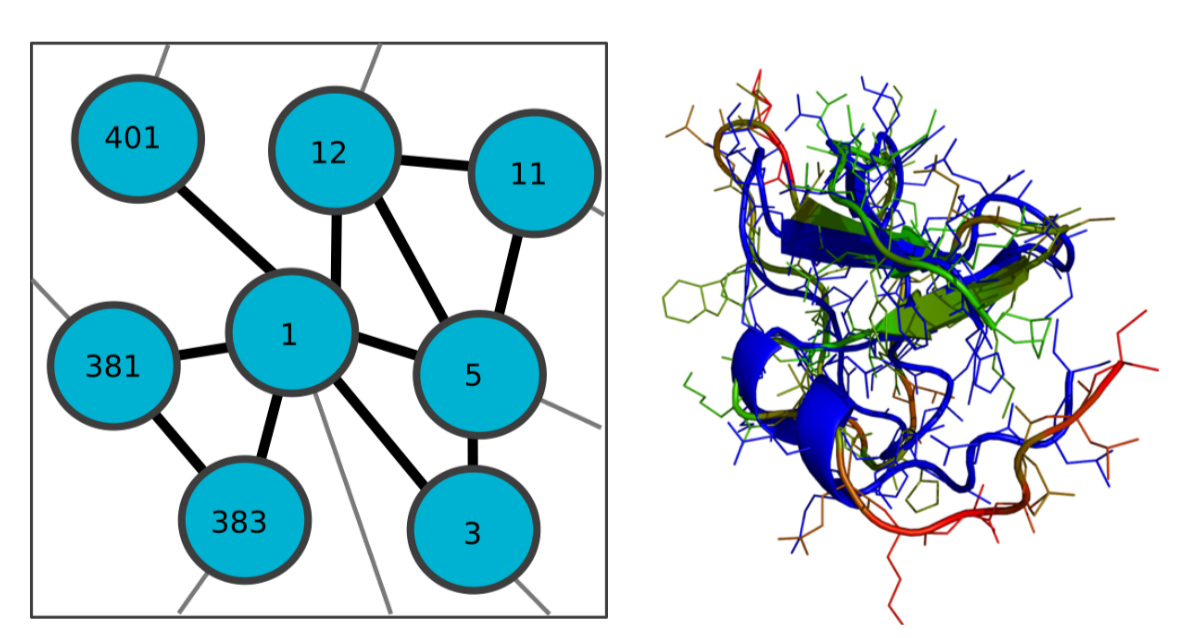
The ongoing advances in sequencing technologies have provided a massive increase in the availability of sequence data. This made it possible to study the patterns of correlated substitution between residues in families of homologous proteins or RNAs and to retrieve structural and stability information. Direct coupling analysis (DCA) infers coevolutionary couplings between pairs of residues indicating their spatial proximity, making such information a valuable input for subsequent structure prediction.
We present pydca, a standalone Python-based software package for the DCA of protein- and RNA-homologous families. It is based on two popular inverse statistical approaches, namely, the mean-field and the pseudo-likelihood maximization and is equipped with a series of functionalities that range from multiple sequence alignment trimming to contact map visualization. Thanks to its efficient implementation, features and user-friendly command line interface, pydca is a modular and easy-to-use tool that can be used by researchers with a wide range of backgrounds.
pydca can be obtained from GitHub or from the Python Package Index under the MIT License.
See also Zerihun, M. B., Pucci, F., Peter, E. K., & Schug, A. (2020). pydca v1. 0: a comprehensive software for Direct Coupling Analysis of RNA and Protein Sequences. Bioinformatics, 36(7), 2264-2265.
propulate
propulate (parallel propagator of populations) is an evolution-inspired hyperparameter optimizer in MPI-parallelized fashion. In order to be more efficient, the generations are less well separated that they often are in evolutionary algorithms. In propulate, a new individual is generated from a pool of currently active and already evaluated individuals that may be from any generation. Individuals may be removed from the breeding population based on different criteria.
propulate can be obtained from GitHub.