
Ohne Proteine geht im Körper nichts – sie sind die molekularen Alleskönner in unseren Zellen. Arbeiten sie nicht richtig, kann das schwere Krankheiten auslösen wie etwa Alzheimer. Um Methoden zu entwickeln, nicht funktionierende Proteine zu reparieren, muss man deren Struktur kennen.
Mit einem Big-Data-Ansatz haben Forscher des Karlsruher Instituts für Technologie (KIT) am Scientific Computing Center eine Methode entwickelt, mit der sie Proteinstrukturen vorhersagen können. Auf Basis statistischer Analysen können auch komplizierte Proteinstrukturen unabhängig vom Experiment vorhergesagt werden. Diese experimentell zu bestimmen wäre dagegen sehr aufwändig und der Erfolg nicht garantiert.
Die Studie wurde in der renommierten Zeitschrift Proceedings of the National Academy of Sciences of the United States of America (PNAS) veröffentlicht (zum Artikel).
Weitere Informationen: Presseinformation des KIT
Forschungsgruppe Multiscale Biomelecular Simulation am SCC
Kontakt am SCC: Dr. Alexander Schug
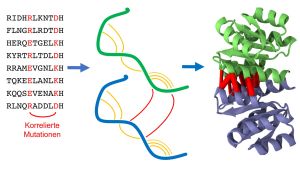
Bild 1: Homodimere sind identische Paare von Eiweißketten (Proteine, grün und blau), die aneinanderbinden. Die statistische Analyse der Proteinsequenzen sucht nach Mutationen, die auf eine räumliche Nähe von Proteinteilen hindeuten, sowohl innerhalb des gleichen Proteins (orange) wie auch mit dem Partnerprotein (rot). Diese Information ermöglicht es, die Proteinstruktur des Homodimers vorherzusagen. (Foto: KIT)